Infectious proteins on the brain: Alzheimer's and prions


Scientists seeking to understand the fundamental pathology of Alzheimer's disease have long debated the merits of two different models of what goes wrong. Is Alzheimer's the result of a widespread, gradually mounting defect in brain biochemistry to which neurons for memory and cognition are particularly vulnerable, like canaries in a coal mine? Or is Alzheimer's caused by a specific, local abnormality that starts in the memory centers and spreads elsewhere, like pollution from a tainted spring?
The widespread defect model has prevailed for the past couple of decades, but some recent research is giving new life to that second, internal infection model. This reevaluation starts to draw together some changes in thinking not just about Alzheimer's but also the prion diseases, like mad cow disease and Creutzfeldt-Jakob, and might even have implications for conditions such as Parkinson's disease and amyotrophic lateral sclerosis (Lou Gehrig's disease).
The travels of tau
At the beginning of February, a report by Li Liu, Scott A. Small, Karen Duff and others in PLoS One grabbed headlines for what it showed about a protein called tau, which is linked to neuron death in Alzheimer's disease. These Columbia University researchers had studied mice genetically engineered to produce the human tau protein in the portion of their brains where the degeneration of Alzheimer's usually starts.
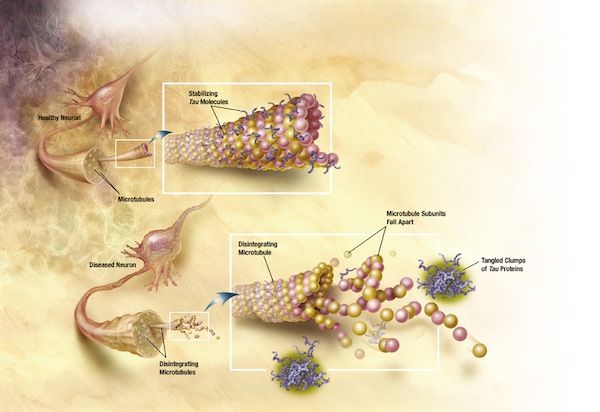
What they found, rather unexpectedly, was that tau started spreading from that area of the brain along the neural connections, precisely mimicking the progression in damage usually associated with the worsening of Alzheimer's disease. Almost like a virus, the tau protein seemed to be moving from one neuron to the next through their synaptic connections. A forthcoming study led by Bradley T. Hyman and his Harvard University team, to appear in the journal Neuron, reportedly arrives at similar conclusions about cell-to-cell transmission of the Alzheimer's degeneration.
(In the interest of heading off panicky misunderstanding, I'll belabor an important distinction: Just because an Alzheimer's-related protein may be weakly infectious within the body does not necessarily mean that Alzheimer's disease is infectious between people. The Columbia and Harvard results don't suggest that people are in danger of catching Alzheimer's disease, only that harmful proteins emerging in one part of the brain can move to other parts. In practice, that fact may bear little on how the disease emerges in the population.)
The tau discovery has several levels of significance -- starting with professional bragging rights but extending on to potential treatments.
Anyone who has followed Alzheimer's disease research knows about the longstanding tension in the field about which abnormal proteins might be most responsible for the condition. In one corner, for example, is Dennis Selkoe of Harvard Medical School and Brigham & Women's Hospital, one of the major proponents of the idea that a protein called beta amyloid is the main villain. Plaques of beta amyloid accumulating around neurons are one of the earliest signs of pathology in Alzheimer's; they are also part of the brain pathology in Down syndrome and some other neurodegenerative disorders. Certain forms of beta amyloid are toxic to neurons. And the infamous prion diseases, such as mad cow disease and kuru, are caused by a renegade amyloid protein, too.
Yet sometimes neurons can be surrounded by plaques and remain perfectly healthy. So in the opposing corner are scientists such as Rudy Castellani of the University of Maryland, who has denounced the beta amyloid hypothesis as "deeply flawed and certainly unproven." Castellani has argued instead that tau, which in Alzheimer's disease forms abnormal tangles inside neurons, is more singularly important.
But because the tau tangles are inside the neurons and not outside them as the plaques are, they have looked like a less tractable therapeutic target. The tangles also show up fairly late in the disease's progression, so much of the Alzheimer's field has often tended to regard tau as delivering a coup de grace to neurons already doomed by beta amyloid plaques.
If the spread of tau tangles from neuron to neuron starts to look like a better trigger for actual pathology, one consequence of the work by Duff and her group might be to swing more attention toward tau and its effects.
Moreover, if molecules of tau are moving between neurons, then they may be at least briefly accessible to, say, antibodies directed against them. Such antibodies could become the basis for Alzheimer's therapies if they truly interrupted the progression of the disease. If nothing else, though, the results out of Columbia suggest that treating Alzheimer's early might indeed help to stem the progression of the disease by containing the degeneration to certain brain centers, before it can spread further.
How prions propagate
None of these developments completely takes beta amyloid off the hook, however. In fact, long simmering science is leading some Alzheimer's researchers to think about whether beta amyloid might have some weakly infectious properties of its own, like an extremely attenuated version of a prion.
To understand how, it's worth first revisiting what prions are and how these simple proteins manage to propagate like viruses. Prions are small, abnormally folded versions of a protein designated PrP. The standard description of how prions reproduce themselves is a study in nature's freakish improbability: the prion supposedly sidles up to a normal PrP and induces it to refold itself into the prion's own abnormal configuration. The two prions then go off to repeat this trick, and the exponentially rising number of prions becomes toxic to the neurons holding them.
That's the model for prion replication first proposed by their discoverer, Nobel laureate Stanley Prusiner, and it's the only one mentioned in most popular science writing. Yet it is not the only possibility, and the others do not rely on quite the same "who'd-a-thunk-it" unlikelihood.
One was originally suggested by Peter Lansbury's experiments at the Massachusetts Institute of Technology in 1993. Small numbers of PrP proteins may occasionally link up to form a short chain, or oligomer. Once that oligomer forms, it avidly latches onto other individual PrP proteins and binds them into a rapidly growing fibril, or filament. Occasionally, however, oligomer-size pieces break off the ends of the growing fibril -- whereupon they form the seeds of new fibrils, and so on.
Another model, in sync with work by Saravanakumar Narayanan and others, is that small, ring-shaped PrP oligomers might act as templates that coax other PrP units into similar rings.
Toxic little chains
These alternative mechanisms involving prion oligomers start to look even more intriguing in light of experiments hinting that free oligomers of beta amyloid -- and not the plaques or individual molecules themselves -- might be harming neurons in Alzheimer's disease.
As Jim Schnabel recently described in a great overview for the Dana Foundation, Konrad Beyreuther of the University of Heidelberg noticed in the early 1990s that beta amyloid forms short oligomers, and William Klein of Northwestern University later demonstrated that these oligomers are directly toxic to neurons in ways that the larger fibrils in the plaques are not. Evidence since then only reinforces the idea that if beta amyloid is what's damaging neurons in Alzheimer's, its oligomers may be the form doing it.
If the alternate mechanisms for prion formation have any relevance, then perhaps the fibrils in the plaques become an ongoing source for beta amyloid oligomers -- maybe by breaking off the ends. And (if we can allow the pile of "ifs" to grow still higher) it's also conceivable that beta amyloid oligomers might pass from one neuron to the next, as Duff's group saw that the tau protein did.
But wait! The situation grows even more complicated (and fascinating). Some studies, such as one from 2010 by Heike Goehler and her colleagues at the Max Delbrück Center for Molecular Medicine in Berlin, hint that different types of amyloid proteins might in some cases be able to trigger one another's aggregation into fibrils and plaques. That is, beta amyloid oligomers might in theory be able to set off the formation of tau tangles, or vice versa.
This "cross talk" between amyloid proteins might not even be restricted to ones involved in a single disease. Work by Claudio Soto's group at the Mitchell Center for Alzheimer's Disease at the University of Texas Health Science Center in Houston found that exposure to prions could accelerate how fast transgenic Alzheimer's-model mice developed amyloid plaques. And as Schnabel notes:
[O]ver the past several years, other experimenters have found evidence that aggregates of tau, huntingtin, alpha-synuclein, and the Lou Gehrig’s disease-associated protein superoxide dismutase 1, all can spread—in prion-like fashion—from cell to cell, in the lab dish or in mouse brains. In recent clinical trials of neuronal transplants, alpha-synuclein aggregates appeared to spread from Parkinson’s patients’ brains into healthy, transplanted neurons.
What to make of all these findings? The science isn't yet settled. At a bare minimum, though, researchers have more reason to look for deep connections or parallels that might run through a number of neurodegenerative diseases, and to hope that any clues bringing a measure of relief or protection against some of them will help with the others, too.
•
Image: PET brain scans. (Credit: Brookhaven National Laboratories)
This post was originally published on Smartplanet.com