MIT’s machine learning designed a COVID-19 vaccine that could cover a lot more people

There are currently 25 vaccines to fight COVID-19 in clinical evaluation, another 139 vaccines in a pre-clinical stage, and many more being researched.
But many of those vaccines, if they are at all successful, might not produce an immune response in portions of the population. That's because some people's bodies will react differently to the materials in the vaccine that are supposed to stimulate virus-fighting T cells.
And so just figuring out how much coverage a vaccine has, meaning, how many people it will stimulate to mount an immune response, is a big part of the vaccine puzzle.
With that challenge in mind, scientists at Massachusetts Institute of Technology on Monday unveiled a machine learning approach that can predict the probability that a particular vaccine design will reach a certain proportion of the population. That doesn't mean they can guarantee its effectiveness, but the scientists' work can aid in knowing up-front whether a given vaccine will have large gaps in who it can help.
The good news is, the MIT scholars have used their approach to design a novel COVID-19 vaccine on the computer that has far better coverage than many of the designs that have been published in the literature this year. They're now testing the design in animals.
The bad news is, there could very well be large gaps in coverage of some of the existing vaccines already being explored by companies and labs, according to one of the authors of the report, David K. Gifford, who is with MIT's Computer Science and Artificial Intelligence Laboratory.
"While they may protect more than 50% of the population, certain individuals and older individuals may not be protected," Gifford told ZDNet in an email, when asked about vaccines currently under trial and in development.
The long path to a vaccine
Vaccines in development were not the direct subject of the work. Most of those vaccines are closed designs; no one knows exactly how they are composed. Instead, Gifford and colleagues designed vaccines from scratch, and then analyzed how effective they are, and extrapolated the findings to a group of vaccines whose composition is known.
Based on that, one can infer there might be problems with vaccines whose exact composition is not known.
It must be borne in mind that any in silico vaccine design such as the kind discussed here is only the beginning of a process that can take years to go through in vivo testing, in animals and then in humans, to establish both safety (non-toxicity), and efficacy, meaning that it actually confers a significant immune response.
Also: MIT's deep learning found an antibiotic for a germ nothing else could kill
But the work shows the ability of large computer models to dramatically speed up the initial work of searching through many, many possible combinations within a universe of possible ingredients, a search that can itself take years at the front end of a drug development pipeline.
This is the latest in large-scale, in-silico efforts against pathogens seen this year from MIT. Back in March, ZDNet reported on how MIT scientists used large-scale machine learning to search many combinations of compounds to come up with a novel antibiotic for a germ nothing else could kill.
A combinatorial challenge
The present work, titled, "Computationally Optimized SARS-CoV-2 MHC Class I and II Vaccine Formulations Predicted to Target Human Haplotype Distributions," is published in Cell Systems, an imprint of Cell Press, part of Elsevier. Authors include Gifford as the corresponding author; lead authors Ge Liu and Brandon Carter of the AI lab; Trenton Bricken of Duke University; Siddhartha Jain, also of the AI lab; and Mathias Viard and Mary Carrington, who have dual roles at Mass General and at the Frederick National Laboratory for Cancer Research in Maryland. (A blog post has also been provided by MIT.)
Gifford and colleagues built a program that designs a vaccine based on two different criteria, the intersection of which is a combinatorial problem.
The first criterion is whether parts of a virus bind to proteins on the surface of a human cell. The bits of virus, which are short strings of perhaps 8 to 25 amino acids, are known as peptides. The human proteins are what are known as surface cell receptors.
When an invading organism enters the body, such as a virus, some of the peptides of that organism fit into a groove in the surface cell receptor. The surface cell receptor then presents that peptide to the body's T cells as a signal of the invasion. The T cells begin a process of killing off such infected cells. That's how natural human immunity works.
But humans need help sometimes, they need to be primed to respond, and that's what vaccines do. Vaccines duplicate this natural immune process before someone is infected, to get the body prepped to generate a T-cell response. To figure out which peptides will fit in the groove of surface cell receptors is a matching problem on a large scale: which of thousands of peptides go with thousands of variants of different surface cell receptors the body can produce.
That's part one. The second part of the problem is asking which people in the population have the combination of alleles, genetic variants of the surface cell receptor, that will work with a certain group of peptides. It's a matter of finding the lowest common denominator in the matching search, which peptide-receptor combinations are common among the greatest number of individuals.
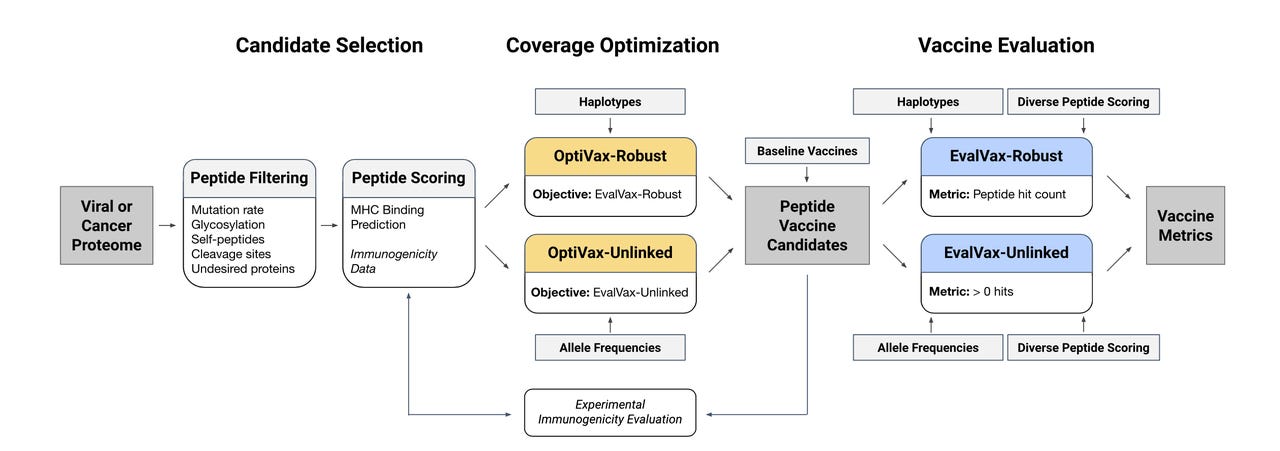
A diagram of the workflow of MIT's machine learning programs for vaccine design. The OptiVax algorithm searches for optimal binding pairs of peptides and human cell surface receptor proteins. It is composed of a novel assembly of eleven existing machine learning search programs. Its objective function is the information about optimal population coverage fed to it by the second algorithm, EvalVax, which analyzes frequency of genetic variants across the population. Two versions of each program options in the workflow, a simpler version called Unliked and a more sophisticated version, known as Robust, which tracks not just single variants in human genes but linked sets of variants known as haplotypes. The option to cover haplotypes is an advanced feature that sets the search apart from past efforts.
An exhaustive search
To achieve both results, the researchers built two machine learning programs. One, called OptiVax, performs the matching search on a scale never achieved before. It combines eleven different pre-existing programs designed to test combinations of peptides and receptors, an ensemble, as Gifford and colleagues call it.
"This is to our knowledge the first application of combinatorial optimization to peptide vaccine design," Gifford told ZDNet, "and it is a challenging computational task that required an efficient implementation."
Just identifying the relevant peptides, about 155,000 in this case, was the first challenge, breaking down the SARS-CoV-2 genetic sequence into its components. Then OptiVax had to go to work on choosing amongst them to pick the best handful, or set, of peptides on which to focus.
"Previous work did not do this exhaustive search," Gifford told ZDNet.
A second program, called EvalVax, takes population data from thousands of individuals who self-reported across three categories, white, Black, and Asian. You could call these ethnicities, and that term is used in the report. Another term that has been proposed in prior work is genetic ancestry. In a 2015 paper, Tesfaye B. Mersha and Tilahun Abebe of the University of Cincinnati proposed ancestry as a better term for genetic distinctions in groups of the population, versus ethnicity, which has more to do with "traditions, lifestyle, diet, and values," they wrote.
A new machine
The two programs, OptiVax and EvalVax, work in tandem in a feedback loop. More specifically, the population program EvalVax, which knows how common alleles are in the three groups of ancestry, serves as the objective function to the search that OptiVax is conducting over peptide-receptor pairs.
All that translates into "about 12 hours on a large multiprocessor computer to design one vaccine using our methods," said Gifford.
OptiVax's ensemble is itself the result of years of prior work by other researchers to create machine learning-based peptide screens. One of the most prominent software programs is called NetMHCPan, developed in 2007 by Morten Nielsen and colleagues at the Technical University of Denmark. NetMHCPan uses a feed-forward neural network. The network is fed pairs of peptide and receptor as its input data, and it generates a predicted binding, or affinity, score, as its output. That score is tested against known bindings that have already been established experimentally, as the supervised training step.
The network's binding predictions are then improved with repeated attempts, via the back-propagation method. Over the years, the program has gone through several revisions and is available as a Web-based server and for download.
The latest in deep learning techniques
The OptiVax program that Gifford and colleagues developed combines NetMHCPan's predictions with predictions from similar screening programs. To get a consensus from the ensemble, OptiVax uses a technique called beam search, which has become ubiquitous in natural language programs. It forms the decoder in software such as Google's BERT and OpenAI's GPT-3. Beam search evaluates a host of possible combinations of elements to find the most likely combo.
To create EvalVax, the objective function that measures population coverage, Gifford and team went beyond the past attempts to measure population coverage. Such studies only asked how common a given genetic variant might be, the allele. But some alleles' frequency can be connected to how common or rare other alleles are, a phenomenon known as linkage disequilibrium.
For that reason, it can be important to look at how common whole combinations of alleles are, known as haplotypes. That, again, brings a combinatorial challenge that is larger, more complex. The approach, however, is a better way to design vaccines, insists Gifford.
"Unlike previous methods, we use HLA haplotype frequencies to score and design vaccines which is a more accurate way of predicting vaccine coverage than the previous use of independent HLA frequencies," said Gifford. HLAs is the technical term for the cell surface receptors that bind with the peptides.
A better vaccine
The result of all this is that OptiVax came up with some vaccine designs consisting of peptide-receptor pairs that have better coverage than designs other teams have come up with since the pandemic started. In what appears reminiscent of many machine learning benchmark tests, the authors report how the coverage of their recipe of peptides compares versus what they estimate to be coverage for the many vaccine proposals in the literature.
In one instance, OptiVax came up with a collection of 19 peptides that would have a 99.91% probability of at least one of the peptides binding to any haplotype of a person in any of the three ancestry groups. That percentage probability of at least one hit was well above the percentage probability for at least one hit in the other vaccine proposals they surveyed from the literature.
As they write in the paper, "We observed superior performance of OptiVax-Robust-designed vaccines on all evaluation metrics at all vaccine sizes […] Most baselines achieved reasonable coverage […] However, many failed to show a high probability of higher hit counts."
A separate question from coverage is how much immunity is conferred by a given vaccine design. With a novel disease like COVID-19, scientists are still finding out which immune responses are neutralizing, meaning, able to retard or stop completely the functioning of the virus.
Happily, there is evidence that peptides that successfully bind to a receptor have a better chance of producing the neutralizing response.
"When peptides do bind to class I MHC molecules, it has been shown in mouse models that almost all binding peptides are immunogenic," Gifford told ZDNet. He was referring to the major histocompatibility complex, the area of the human genome that produces the receptors. A study in 2018 by Washington University researchers, Gifford noted, found that "a surprisingly high fraction" of such peptides produced a neutralizing response, what's known as immunogenicity.
In contrast, many drugs in development may miss the mark in neutralization even if they stimulate some response, Gifford cautions.
"While it is early days, clinical study data on candidate vaccines that has been released has shown that not all individuals develop a robust cellular immune response to COVID-19." Gifford speculates that, as EvalVax suggests, those vaccines "have population coverage gaps in peptide binding," which, he said, "could influence durability and response in older individuals."
A complicating factor is that COVID-19 continues to evolve genetically, so that some proteins change over time, making it harder to target the peptides they contain.
The mutation rate appears to be small enough at present not to be a substantial issue, Gifford told ZDNet.
"We can not guarantee that there will not be further viral sequence drift," said Gifford. "However, the lack of, or low rate of mutation of our candidate peptides over our more than 4,000 geographically-sampled genomes suggests that these peptides may be functionally required and thus less likely to drift in the future."
Ancestries challenge coverage
One nagging problem persists despite the significant improvement observed in OptiVax's design: even with better overall coverage, some of the results are mixed according to ancestry.
When comparing vaccine designs from OptiVax in terms of having two or more peptide hits, the percentage probability declines for all three ancestry groups, but it declines unevenly. The likelihood of having multiple hits starts to show great disparity at five or more hits, with those of Asian ancestry showing the greatest likelihood of the full number of hits, those of white ancestry showing somewhat less, and those self-identifying as of Black ancestry showing less of a chance than either two.
That's important, because any one peptide-receptor hit might not turn out to be effective in a given individual, so it's better, if possible, to have multiple potential peptides to increase the odds a drug works on a given individual.
It's tempting to think that adjusting the search techniques could reduce that disparity. In the present paper, Gifford and colleagues focused on what's called precision, meaning, making sure that there are as few as possible false positives in their peptide selections. Because drugs can take a long time to develop, a lot of effort can be wasted if early positive indicators later turn out to have been misleading.
Also: How a smartphone coupled with machine learning may become a simple, efficient test for COVID-19
But focusing on precision in this case meant less emphasis on what's called recall, which is the number of true positives found out of all the true positives that exist in a universe of possibilities. It would be nice to think that adding more focus to recall could lead to more peptides that would bind across the haplotypes of all three ancestries, or at least, more evenly so.
That may not be the case, however.
"Our estimates of self-reporting ancestry-based haplotype populations suggests that certain ancestries may be intrinsically harder to cover," Gifford told ZDNet when asked about increasing recall. In fact, greater recall could be misleading, as it "would make the numbers look better, but it would potentially overestimate population coverage and as a consequence provide less robust vaccine designs."
Gifford cautions not to read too much into the disparities at high hit counts, as those disparities can be a result of "many factors."
Gauging COVID-19's risk
In the meantime, there is ongoing work to take the OptiVax design to the next level. "We are working with both academic and commercial collaborators to test OptiVax-derived designs in animal models," Gifford told ZDNet. "If the designs show promise in these models, the next logical step would be clinical trials."
Beyond just drug development, this kind of combinatorial analysis can pay a lot of other dividends. A separate project of the authors is currently underway examining the blood sera of people who have recovered from COVID-19 to gauge how much immunity those individuals developed.
One might wonder, too, if the OptiVax and EvalVax findings reveal anything about the pathogenesis of COVID-19. Is there anything that can be said about the different ancestries' peptide binding rates that reflects upon those populations' response to the disease?
It turns out that Gifford and team have also added that question to their work. They are comparing the patterns of peptide and receptor matches they have found to patterns of COVID-19 severity in patients, accompanied by analysis of healthy control subjects, to do risk analysis of the disease, Gifford told ZDNet.
Vaccine makers should open up
The authors have some choice words for those developing vaccines. Consistent with the warning that Gifford gave ZDNet in email, the concluding section of the published paper notes a big potential problem with many vaccines. They tend to focus a lot on the most notorious protein of the SARS-CoV-2 virus, called the S, or spike, protein. There is a good reason vaccine development has focused on the S protein, and it's because biological analysis suggests the S protein should produce antibodies that are of the neutralizing kind.
But the OptiVax test suggests the S protein may not have complete coverage of the population. "Vaccines that only employ the S protein may require additional peptide components for reliable CD4+ T cell activation across the entire population," the authors write. They suggest ways of adding peptides to S-based drugs to enhance coverage.
On a deeper note, Gifford and team urge drug developers to put their designs out in the open to be scrutinized. "The precise designs of most of these vaccines are not public," they note. "We encourage the early publication of vaccine designs to enable collaboration and rapid progress toward safe and effective vaccines for COVID-19."
The OptiVax code and data sets of its peptide predictions are available on Github.