With unprecedented amounts of genetic data, researchers are tracking how COVID-19 mutates around the world

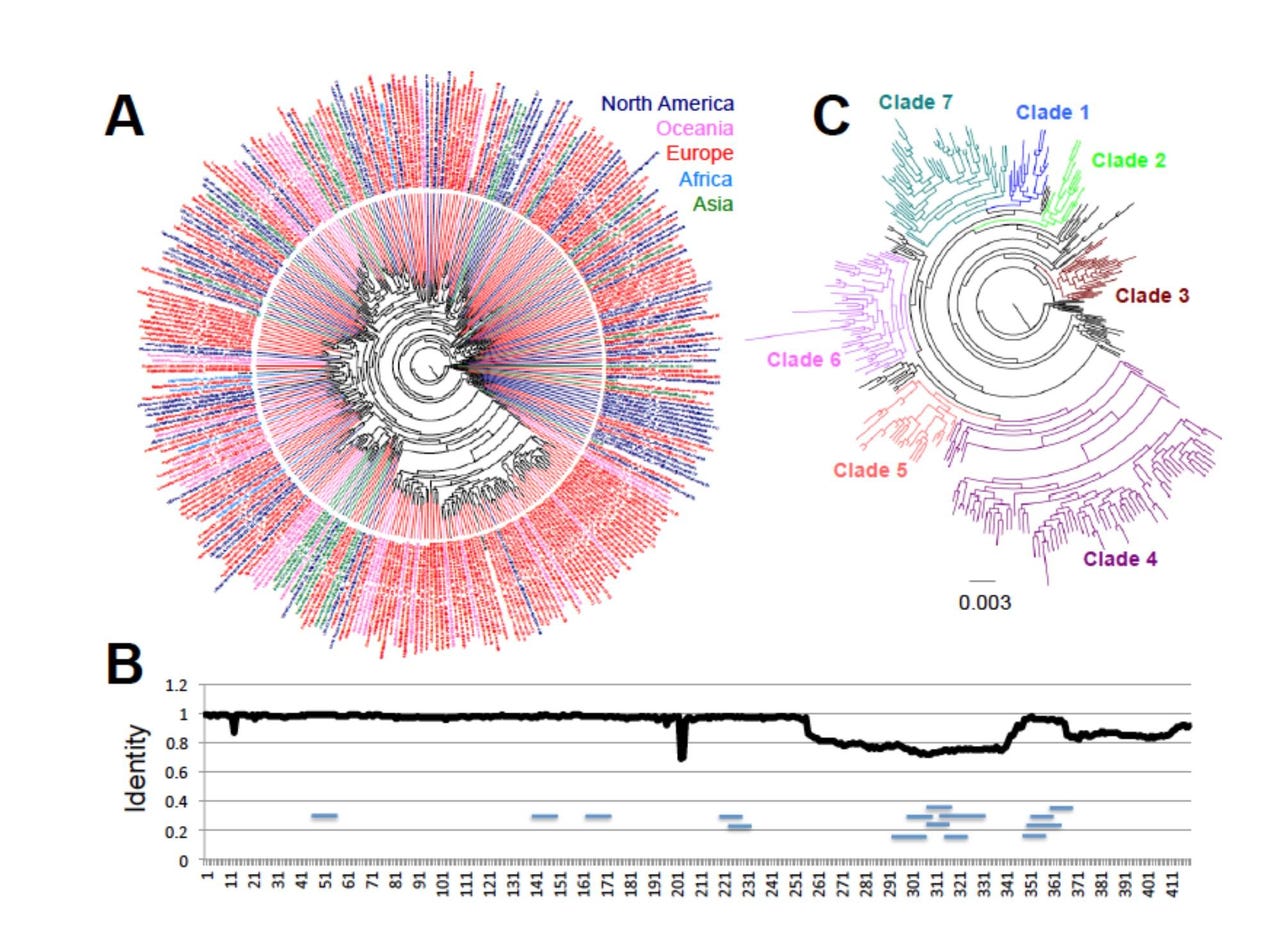
A view of the phylogeny of the N protein of the SARS-CoV-2 coronavirus, one of the key parts of the virus, seen as diverging versions, or sequences, that are spread throughout the world. Dumonteil and Herrera use smart software that can make statistical inferences about how one sequence relates to another and therefore how sequences are evolving from the original form of the virus.
The world has been obsessed with surveillance of a particular kind for six months: watching people to see who's sick.
There is another form of surveillance that is just as important but less well understood, and that is the attempt to track how the SARS-CoV-2 virus itself is changing as it spreads around the world.
COVID-19 , like other coronaviruses, evolves rapidly. The particular order of nucleic acids encoded in its RNA, the genetic instructions that create the proteins that make up the virus, change over time. (Humans have a double-stranded DNA, whereas viruses, for evolutionary reasons, have the single-stranded genetic material RNA.)
That rapid change in RNA is a problem for efforts to counter the virus that depend on knowing what the virus looks like at a molecular level. The many vaccines in development, for example, will only work if they're tuned to the proper sequence of amino acids that are in the virus in its current form. If the virus that's circulating among people suddenly morphs a different sequence, it could render a vaccine useless.
The same is true for serologic tests that measure the presence of antibodies in people who've had the disease. They work by testing blood by presenting a small bit of the virus's genetic information, known as the antigen, to see if anything in the blood responds to that antigen. If a response is seen, that means there are antibodies. Hence, such tests won't work if the antigen, the piece of virus protein, changes form.
So scientists are trying to develop an ongoing family tree that records how the virus changes form.
Scientists Eric Dumonteil and Claudia Herrera of the School of Public Health and Tropical Medicine at Tulane University this month described their attempts to build such a family tree using 18,247 samples of the viral RNA, what they refer to as "a global analysis of viral diversity across the world."
Their paper, "Polymorphism and selection pressure of SARS-CoV-2 vaccine and diagnostic antigens: implications for immune evasion and serologic diagnostic performance," was posted June 18th on the bioRxiv pre-print server. The work has not been reviewed yet by peer researchers, and so its findings have to be taken with great caution.
The samples of COVID-19 RNA can be downloaded as files from GISAID, a database hosted by Germany that is drawn upon by scientists all over the world. The U.S. Centers for Disease Control has been a technical partner supporting the database since its creation in 2006.
To see how those thousands of samples relate to one another, Dumonteil and Herrera turned to a software package called FastTree, developed by Morgan N. Price and colleagues at Lawrence Berkeley National Laboratory in 2009.
FastTree is designed to infer phylogenies, which means to calculate how close one sample of DNA or RNA is to another to the extent that it could have evolved from it by some change of one or more nucleic acids. To do that with thousands of samples of genetic material is a large combinatorial problem that can quickly get out of hand in terms of computing power required. Just the storage in computer memory needed to hold the values of all those examples quickly rises into the tens of gigabytes.
So FastTree takes some shortcuts, such as grouping together samples of RNA as profiles that summarize how any two differ from one another. It's a kind of data compression, if you will, that makes it easier to search and compare the large collections of RNA.
Once the phylogenies, or family trees, are constructed with FastTree, a second program, HyPhy, is used to test out various hypotheses about how one sample of RNA may have evolved from another. The software was first introduced in 2000 by statistician Spencer Muse of North Carolina State University and biologist Sergei Kosakofsky Pond of Temple University.
The conclusion that Dumonteil and Herrera arrive at is that COVID-19 is "a fast evolving virus, as it is rapidly accumulating mutations." And it's not just changes in a particular area of RNA, it's widespread. The changes in nucleic acids are "scattered through the viral genome, rather than clustered in specific genes," the authors write. Several new "clades" have been emerging, where a clade is a cluster of examples that share a common parent from which they sprung. It's like a new colony of settlers setting up in a new land.
The scientists are seeing evolutionary pressure at work as the virus seeks to evade the body's immune system. Like every organism, natural selection means that as mutations happen in the virus, some will come to dominate because they help it better survive in any number of ways, whether it's attaching to the human cell better or replicating better.
ZDNet reached out to Dumonteil in email to ask follow-up questions.
Even by the standards of rapidly evolving coronaviruses, the number of mutations seemed substantial, Dumonteil indicated to ZDNet.
"The size of the pandemic has also allowed for a considerable number of virus generations within a relatively short time, so these changes may also reflect virus ongoing adaptation to a new host (humans)," Dumonteil told ZDNet by email.
"The selection pressure we detected is part of this process and it does reflect how our immune system is attempting to control the virus," he said.
What's less clear at this point is whether the number of rapid mutations means humans are presenting a formidable challenge or if, on the contrary, the virus is doing so well that it has more opportunity to evolve. It's hard to make definitive conclusions because the scale of the data introduces statistical uncertainty. The rapidity of mutation could simply be a result of the abundance of viral samples gathered.
"The percentage of variability in these antigens from SARS-COV-2 seems high compared to other RNA viruses, but this may be due to the unprecedented level of sequence data available," Dumonteil told ZDNet.
Dumonteil said it's not clear yet from the lineages themselves how successful humans have been at fighting the virus. To draw any conclusion would be going beyond what the data show.
For the time being, the key parts of the virus that a serologic test or a vaccine would aim for are well conserved, meaning, they're not changing as much as they might. For example, what's called the S protein, part of which attaches to proteins on the surface of a human cell to gain entry to the cell and replicate the virus, is "highly conserved." For that reason, vaccines or tests looking for the distinct form of the S protein should work.
But ongoing surveillance will be necessary because Dumonteil and Herrera can already see changes in the S protein that are making it diverge from what was seen with the earliest samples of the virus from China. "Most of these variants appeared in the past weeks/months and may be slowly replacing the virus presenting sequences similar to that of the initial isolates from Wuhan, China," they write in the report.
It's a matter of keeping on top of things, Dumonteil told ZDNet. "We are certainly interested in following these changes over time as new sequences become available, as this will allow to adjust both diagnostic tests and vaccine candidates."
Oliver Pybus and colleagues at The University of Oxford used the phylogenies to track by what geographic routes the virus came into the U.K., a kind of genetic passport.
Such analysis is going on all over the world, and it turns up different findings in the hands of different researchers.
For example, Oliver Pybus and colleagues at Oxford this month described how the virus changed form in samples observed in the United Kingdom alone. They used the phylogenies to track where the virus came from geographically like a kind of genetic passport.
By constructing phylogenetic trees, they could infer how much of COVID-19 in the U.K. came from foreign travelers who entered the country in March before global travel bans went into effect. A third of the COVID-19 infection cases may have been imported from Spain, they estimate, another third from France, and the balance from Italy and other countries.
Again, a lot of caution has to be exercised because such knowledge is constructed from statistical tools and is only an approximation of what may have transpired. Nevertheless, Dumonteil's use of the word "unprecedented" to characterize the scale of these projects is worth lingering on. The kinds of viral surveillance going on may yield a scientific picture of infection around the world that is unlike any picture of disease humanity has ever constructed before.